
Podłoże molekularne i potencjalne terapie
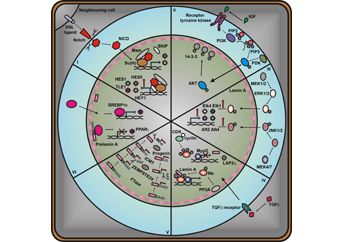
Mimo iż laminy ekspresjonowane są we wszystkich komórkach organizmu, defekty w genach je kodujących powodują bardzo szerokie spektrum objawów, które w zależności od rodzaju mutacji odnosi się do wybranego typu tkanki lub dotyczy funkcjonowania całego organizmu. Do tej pory powstało kilka hipotez wyjaśniających opisywane zjawisko. Dwie najpopularniejsze to: hipoteza stresu mechanicznego i hipoteza ekspresji genów. Pierwsza, bierze pod uwagę funkcje lamin jako białek strukturalnych. Mutacje w genach kodujących laminy, mogą powodować osłabienie prawidłowej struktury blaszki jądrowej i otoczki jądrowej, która staje się w konsekwencji mniej odporna na działanie stresu mechanicznego. Dlatego w tkankach narażonych na działanie sił mechanicznych, takich jak tkanka mięśniowa, ekspresjonujących nieprawidłowe formy lamin dochodzi do deformacji otoczki jądrowej i uszkodzenia materiału genetycznego (charakterystyczne morfologie jąder obserwowano u pacjentów dotkniętych laminopatiami). Dodatkowo potwierdzono, że laminy oddziałują z elementami kompleksu LINC (linker of nucleosceleton and cytoskeleton), stanowiącego miejsce interakcji szkieletu jądrowego z cytoszkieletem. Zaburzenia tego oddziaływania, będące efektem mutacji lamin, mogą powodować nieprawidłową lokalizację jąder komórkowych w obrębie komórki lub zaburzać proces mechanotransdukcji. Druga hipoteza zwraca uwagę na rolę lamin w kontroli ekspresji genów. Laminy uczestniczą w regulacji transkrypcji na różnych poziomach, począwszy od bezpośrednich interakcji z czynnikami transkrypcyjnymi (np. SREBP-1, MOK-2, c-Fos, pRb, Oct-1), poprzez kontrolę struktury chromatyny, stanowiąc miejsce zakotwiczenia domen chromatynowych, po indukcję epigenetycznych modyfikacji histonów (w komórkach pacjentki cierpiącej na progerię Hutchinsona-Gilforda, u której ekspresjonowany jest zmutowany gen laminy A, obserwowano usunięcie z nieaktywnego chromosomu X markera blokującego transkrypcję (H3K27me3) oraz obniżenie aktywności metylotransferazy EZH2, enzymu odpowiedzialnego za wspomnianą modyfikację).
Molekularne podłoże laminopatii typu dystrofii mięśniowych może być bardzo różnorodne. Do tej pory obserwowano zaburzenia funkcjonowania szlaków MAPK, pRb, MyoD, WNT/β katenina oraz TGFβ.
Projekty i aktualności:
Projekt finansowany ze środków Wrocławskiego Centrum Badań EIT+ w ramach realizacji projektu 'Biotechnologia i zaawansowane technologie medyczne' - BioMed (POIG.01.01.02-02-003/08) finansowanego ze środków Europejskiego Funduszu Rozwoju Regionalnego (Program Operacyjny Innowacyjna Gospodarka, Poddziałanie 1.1.2).
