
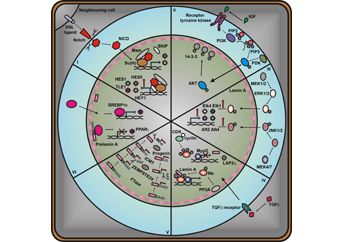
PROPONOWANA TERAPIA
Do tej pory nie opracowano skutecznej terapii, która mogłaby być zastosowania u chorych dotkniętych laminopatiami. Leczenie jest głównie leczeniem objawowym. W przypadku laminopatii typu dystrofii mięśniowych bardzo ważne jest wczesne, prawidłowe rozpoznanie choroby i systematyczne monitorowanie czynności serca, gdyż zawały stanowią główną przyczynę śmierci pacjentów. Chorym wprowadza się rozrusznik serca, wykonuje przeszczepy serca, poddaje się zabiegom rehabilitacyjnym związanym z występującymi przykurczami. Wydaje się, ze nowym podejściem terapeutycznym zastosowanym u pacjentów może być wykorzystanie inhibitorów szlaku kinaz aktywowanych miotogenem. Specyficzne inhibitory hamujące aktywność ERK i JNK u dystroficznych myszy spowodowały wyraźną poprawę fenotypów zwierząt, zwłaszcza dotyczącą funkcjonowania serca. Ponieważ zawały stanowią główną przyczynę zgonów pacjentów dotkniętych laminopatiamii, zastosowanie omawianych inhibitorów, wydaje się być bardzo korzystne [Muchir i wsp., 2009 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2638780/, Wu i wsp., 2010 http://www.ncbi.nlm.nih.gov/pubmed/20388542].
W przypadku laminopatii związanych z przedwczesnym starzeniem się, wielkie nadzieje wiąże się z zastosowaniem inhibitorów transferazy farnezylowej (FIT) takich jak lonafarnib lub tipifarnib, substancji wykorzystywanych w czasie terapii pacjentów z chorobą nowotworową. Związki te blokują proces farnezylacji prelaminy A. Ich zastosowanie w hodowlach komórkowych przyniosło bardzo dobre efekty (wyraźna poprawa morfologii jader komórkowych), również u transgenicznych myszy obserwowano polepszenie fenotypów. Pomimo dobrych efektów wydaje się, że stasowanie związków tego typu ma pewne ograniczenia i może powodować efekty uboczne. Zaobserwowano, że zablokowanie farnezylacji progeryny i prelaminy A może powodować alternatywną modyfikację przy udziale transferazy geranylogeranylowej, co częściowo tłumaczy słabsze efekty obserwowane w przypadku terapii testowanej na mysich modelach progerii. Dlatego w celu wydajnego zablokowania prenylacji próbowano stosować kombinacje statyn i aminobisfosfonianów [Varela i wsp., 2008 http://www.ncbi.nlm.nih.gov/pubmed/18587406]. Pamiętać jednak należy, że zastosowanie inhibitorów farnezylacji zablokuje modyfikacje nie tylko prelaminy A, ale również innych białek (ok. 100 u człowieka, między innymi lamina B, białko Ras, czy regulujące przebieg cyklu komórkowego białko Abl). Poza tym nie możemy wykluczyć, że obecność w komórce niemodyfikowanej progeryny będzie nieszkodliwa. Eksperymenty polegające na ekspresji zmienionej, nie podlegającej fosforylacji progeryny wykazały, iż obecność takiej formy laminy (gdy brak laminy C) przyczynia się do rozwoju kardiomiopatii [Davies i wsp., 2010 http://www.ncbi.nlm.nih.gov/pubmed/20421363]. Pewnych informacji dostarczają również efekty terapii z zastosowaniem inhibitorów proteazy HIV, (hamującej również aktywność ZEMPSTE24) u pacjentów dotkniętych zespołem nabytego braku odporności (AIDS), u których obserwowano pojawienie się lipodystrofii, co wiązane jest z nieprawidłową modyfikacją laminy A [Caron i wsp., 2007 http://www.ncbi.nlm.nih.gov/pubmed/17612587]. W leczeniu progerii, ale również innych laminopatii, gdzie ważne jest zablokowanie ekspresji nieprawidłowej formy laminy, przydatne może się okazać zastosowanie antysensownych oligonukleotydów i shRNA [Huang i wsp., 2005 http://www.ncbi.nlm.nih.gov/pubmed/16208517, Fong i wsp., http://www.ncbi.nlm.nih.gov/pubmed/16511604]. Na razie testy przeprowadzano jedynie na hodowlach komórkowych, jednak przyniosły one bardzo dobre rezultaty.
W leczeniu laminopatii związanych z lipodystrofią stosuje się leki wykorzystywane w terapii uogólnionych lipodystrofii i cukrzycy typu 2, takie jak leptyna i pochodne tiazolidynodionu (glitazony, TZD), natomiast defektom kosmetycznym zapobiega się stosując operacje plastyczne. Wykorzystanie u pacjentów z FPLD (rodzinna dystrofia typu Dunnigana) rekombinowanej leptyny (R-metHuLeptin) powoduje zwiększenie wrażliwości na insulinę, polepszenie metabolizmu glukozy i obniżenie poziomu trójglicerydów. Obserwowany efekt jest jednak słabszy w porównaniu z chorymi cierpiącymi na GL (uogólniona lipodystrofia) [Park i wsp., 2007 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2595136/]. Badania dotyczące podłoża molekularnego laminopatii typu dystrofii mięśniowych wykazały, że mutacje lamin wpływają na zahamowanie aktywności białka PPARγ, dlatego zasadne wydaje się zastosowanie syntetycznych koaktywatorów PPARγ jakimi są glitazony. Substancje te testowano na razie jedynie na hodowlach komórkowych, ale uzyskano spodziewane rezultaty, co stwarza możliwość dalszego ich testowania [Maraldi i wsp. 2008, http://www.ncbi.nlm.nih.gov/pubmed/18155670 ]
Projekty i aktualności:
Projekt finansowany ze środków Wrocławskiego Centrum Badań EIT+ w ramach realizacji projektu 'Biotechnologia i zaawansowane technologie medyczne' - BioMed (POIG.01.01.02-02-003/08) finansowanego ze środków Europejskiego Funduszu Rozwoju Regionalnego (Program Operacyjny Innowacyjna Gospodarka, Poddziałanie 1.1.2).
